Genetics, Pathology, & Potential Future Advances in the Study of Huntington's Disease4. muHtt And Its Role In The Pathology of Huntington’s DiseaseAs wtHtt is so clearly involved in multiple different cellular processes, it is no wonder that a mutation in the protein results in the catastrophic symptoms seen in HD. The aberrant protein causes massive neuronal loss, mainly in the medium spiny neurons of the striatum and the disease progresses to affect the rest of the basal ganglia, before finally affecting other regions of the brain such as the cortex. Much research has been conducted into the pathogenic role of muHtt, with the major question concerned regarding whether the symptoms are caused by a loss of function of wtHtt, or a gain of function of muHtt. 4.1 Huntingtin Aggregates and Inclusion BodiesEarly research into the effects of muHtt used immunostaining to detect where the protein was localised. It was quickly shown that in both mouse models of HD and brains of sufferers, nuclear inclusion bodies were highly prevalent (DiFiglia, et al., 1997). This same research also showed that the inclusion bodies had been targeted by ubiquitin, but had not been removed, suggesting that they had some form of protective mechanism against targeted degradation. This formation of inclusion bodies is a common feature of other neurodegenerative diseases, such as spinobulbar muscular atrophy (Zoghby & Orr, 2000). However, one crucial factor about the inclusions is that despite their association with disease progression, they are not associated with neurodegeneration (Slow, et al., 2005) and they have a protective effect against toxicity in cell cultures (Arrasate, Mitra, Schweitzer, Segal, & Finkbeiner, 2004). This suggests that it is diffuse muHtt that is responsible for cell death and that inclusion bodies may be a protective cellular function.The inclusion bodies are formed of aggregated misfolded N-terminal fragments (DiFiglia, et al., 1997). The fragments aggregate because of propensity of hydrophobic regions to clump together. The polyQ repeat of muHtt exposes normally shielded hydrophobic areas. These fragments are likely formed as the cell attempts the proteolysis of Htt- the protein has a number of protease cleavage sites and the expansion in the polyQ region could be responsible for preventing fragment clearance after protein degradation. Indeed, proteolysis of Htt in mouse models of HD generates large levels of N termini (Zhou, et al., 2003). In this same mouse model, the fragments formed inclusion bodies that were present before the onset of symptoms and this formation was attributed to decreased activity of the proteasome-chaperone system responsible for clearance of misfolded peptides. This would mean that for inclusion bodies to be protective, their formation would effectively be a ‘back-up’ system after the failure of the primary mechanism. Small N-terminal fragments have greater cytotoxicity than longer Htt fragments. Mice that express the small exon 1 fragment of Htt (67 amino acids) with a 115-150Q repeat expansion have greater levels of neurodegeneration than mice solely expressing full length muHtt or even a longer, N171 fragment with an 82Q repeat unit (Li & Li, 2006). However, no protease has been found that cleaves muHtt to release exon 1, here the mice were transfected with synthesised cDNA. Instead, specific caspase cleavage of muHtt is required to form the toxic N-terminal fragment. In vivo studies of inhibition of specific caspases showed that inhibition of caspase 6 but not caspase 3 resulted in no loss of neurons or striatal volume, whereas caspase 6 cleavage of muHtt produced a toxic, 586 amino acid fragment (Graham, et al., 2006). In this study, mice with inhibited caspase 6 not only showed no neurodegeneration, but showed no behavioural changes, which the authors concluded meant that this 586 amino acid caspase 6 fragment of muHtt has a rate limiting role in progression of HD. The key point from this is that not all fragments are toxic, suggesting potential therapeutic roles. Aggregates sequester a molecule called mammalian target of rapamycin (mTOR), a protein required to regulate cell growth, proliferation and protein transcription (Ravikumar, et al., 2004). By sequestering this and subsequently lowering its cellular levels, the aggregates induce autophagy, or controlled cell death. However, this only works for micro-aggregates and larger aggregates are unable to be cleared by autophagosomes (Ravikumar, et al., 2004). The authors suggested that this explained the late onset of HD- eventually production of aggregates will exceed their clearance and symptoms will start to appear. The interesting point about this clearance mechanism is that it does not comprehensively state whether aggregates are toxic or protective. While they do induce autophagy (clearly a protective mechanism to destroy toxic soluble protein), this eventually fails and the subsequent aggregate build up may also contribute to toxicity. While research suggests potential protective roles for cytoplasmic aggregates and inclusion bodies, other evidence points towards their toxicity. In vivo mouse models of inclusion body formation show that the aggregates are associated with the early neuropathology of HD (Li H. , Li, Yu, Shelbourne, & Li, 2001). This disagrees with the research by Ravikumar et al (2004) that the aggregates are actually responsible for the prevention of early HD symptoms. Additionally, it was shown that decreasing inclusion body formation in vitro through over-expression of chaperones and heat shock proteins was associated with decreased cell death (Carmichael, et al., 2000). Yang et al. (2002) introduced small aggregated polyQ peptides to cells in culture to determine their effect and found that any aggregates that moved to the nucleus caused dramatic cell death. Aggregate formation and inclusion bodies are a key feature of HD but there is plenty of conflicting evidence about their role in disease progression. Current research suggests that while aggregates do contribute to cellular toxicity, their role in cell death may be less dramatic than un-aggregated toxic fragments of muHtt. The role of aggregates in inducing autophagy also suggests that their formation is a controlled cellular process. Therefore, it is possible aggregate formation is a compromise- while aggregates may be toxic to cells, formation lowers levels of the more toxic free fragments, maintaining healthy cells and preventing more catastrophic damage that would be associated with uncontrolled cell death. 4.2 Metabolic Dysfunction in Huntington’s DiseaseVery recent research into the toxicity of the N –terminal fragments has shown that they have an effect on mitochondria. Intra cellular transport of mitochondria occurs for a number of reasons, including passing on the organelles to daughter cells and transporting the organelles to areas of the cell with high energy consumption (Saxton & Hollenbeck, 2012). Impairment of this transport has been linked to the development of HD and it has been shown that the soluble, rather than aggregated form of muHtt fragments have been implicated through the known interactions of Htt with transport proteins (Tian, et al., 2014). Prevention of transport may lead to heightened glutamate excitotoxicity and cell death. Excitotoxicity occurs when cells are exposed to dangerous levels of Ca2+ ions as a result of excessive glutamate receptor stimulation. Calcium enters cells through ion channels opened by neurotransmitters but its role in activating enzymes means if levels are too high (mitochondria act as a calcium buffer), dangerous enzymes are activated. If mitochondria are impaired, cells are even more susceptible. Impairment of normal mitochondrial function has profound effects. Mitochondria, as well as their role in producing ATP, have functions including maintenance of the cell cycle, initiating apoptosis and controlling cell signalling (McBride, Neuspiel, & Wasiak, 2006). Mitochondria are a major source of reactive oxygen species (ROS) which can cause oxidative stress damage. Because of this, the organelles contain enzymes essential for dealing with these ROS but if this system was compromised then the species could cause oxidative damage to the mitochondrial membrane, as well as cellular lipid and DNA. Crucially, damaging the mitochondrial membrane could lead to release of cytochrome C, resulting in the induction of apoptosis and suggesting a mechanism for neuronal death.
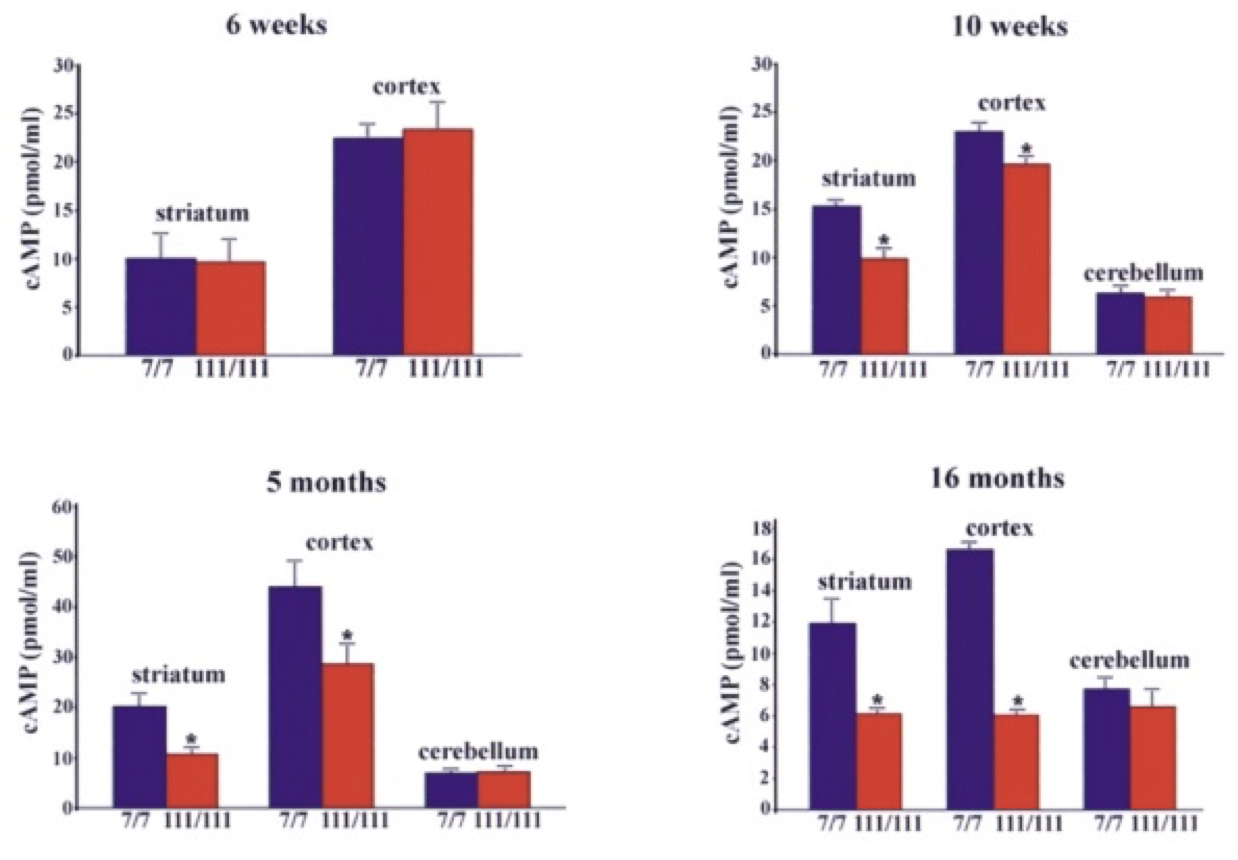
The initial link between mitochondria and HD came from post-mortem analysis of patient striatum, whereupon it was shown the cellular mitochondria had decreased in vitro activity, specifically in protein complexes II and III (Gu, et al., 1996). Studies of Hdh Q111 cells (homozygous for muHtt) showed decreased levels of ATP production (measuring cAMP levels, requires ATP for synthesis) than Q7 cells expressing wtHtt (Gines, et al., 2003), a finding further supported by follow-up studies (Milakovic & Johnson, 2005). This is a progressive reduction (Fig. 5). As well as ATP impairment, lymphoblast mitochondria in HD patients show a proportional decrease in membrane potential with cellular muHtt levels (Panov, Lund, & Greenamyre, 2005). This results in their depolarising at lower cytoplasmic calcium concentrations, thereby increasing their sensitivity to calcium induced cytochrome C release. In striatal cells, muHtt directly interacts with the mitochondrial membrane to induce mitochondrial permeability transition pore opening, followed by a significant release of cytochrome C (Choo, Johnson, MacDonald, Detloff, & Lesort, 2004). This interaction would go some way to explaining the massive cell loss seen in HD. Mitochondrial dysfunction as a result of direct muHtt interactions would also cause an increase in ROS levels. This would have two major effects within the cell. ROS directly damages DNA and analysis of the parietal cortex in post-mortem HD brains show increased levels of 8-hydroxy-2’-deoxyguanosine (8-OHdG), a biomarker for oxidative DNA damage, in mtDNA (Polidori, Mecocci, Browne, Senin, & Beal, 1999). This would further compound the effects of muHtt on mitochondria leading to further metabolic problems. Oxidative damage is also raised in cDNA in the frontal cortex and caudate nucleus (Browne, et al., 1997). This damage is mostly localised to neurons because brain mitochondria are composed of lipids with a higher amount of polyunsaturated acyl groups, a feature that makes them more susceptible to oxidation. Damage to DNA would also lead to mutations and potential increases in the CAG repeat length. Oxidative damage is repaired by the BER system, which can lengthen repeat units (Kovtun, et al., 2007). The second effect of elevated ROS levels is the possible formation of protein carbonyls, molecules which are dysfunctional due to a loss of structural integrity. Increased levels of these are seen in the striatum of mouse models of HD (Perluigi, et al., 2005). Modified proteins include creatine kinase and heat-shock protein 90, proteins where dysfunction would have critical roles in cellular dysfunction. 4.3 Impairment of the Ubiquitin-Proteasome SystemThe ubiquitin-proteasome system (UPS) is responsible for the degradation of cellular proteins, as well as cell signalling and other roles (Imarisio, et al., 2008). Proteins are targeted for degradation by the addition of ubiquitin, a process that requires three enzymes. This tag is recognised by the 26S proteasome, a complex formed of two regulatory 19S subunits and a 20S catalytic subunit. The tagged protein is passed through the 20S core and deactivated by cleavage into small fragments. Crucially, the 20S core can only effectively deal with unfolded protein. The choking hypothesis of UPS impairment in HD states that the expanded polyQ region of the muHtt protein gets ‘stuck’ within the catalytic core of the proteasome. Eukaryotic proteasomes cannot cleave within a polyQ region of between 10 and 30 residues (Venkatraman, Wetzel, Tanaka, Nukina, & Goldberg, 2004). If the polyQ region is less than 30 residues long, it is released intact, whereupon it likely aggregates. Venkatraman et al (2004) predicted that polyQ regions of more than 30 residues may form β turn structures with the internal region of the proteasome, preventing fragment release. This would result in the build-up of muHtt and its subsequent aggregation, as well as the impairment of crucial processes such as the cell cycle. The fact that inclusion bodies contain ubiquitin supports this theory- the protein is not removed from the fragments after failed proteolysis (DiFiglia, et al., 1997). Interestingly, synthetically generated proteins with expanded polyQ repeats do not inhibit proteasome function in vitro, whereas muHtt purified from post mortem HD brains does show in vitro inhibition (Bennett, Bence, & Kopito, 2005)(Diaz-Hernandez, et al., 2006). This means that while it is the polyQ region causing the problem, some other component present in the Htt protein but not in a synthetic peptide is needed for complete inhibition. While some research shows impairment of the UPS, other research shows no change in activity. The rate of degradation of GFP-labelled polyQ peptides in in vitro cultures of SH-SY5Y cells expressing muHtt was no different to that of cells expressing wtHtt (Dinq, et al., 2002). Equally, an increase in proteasome activity has been noted in mouse models of HD (Diaz-Hernandez, et al., 2003), attributed to an increase in levels of proteasome components in the cell, suggesting increased cellular clearance of aberrant muHtt. Clearly, there is conflicting evidence regarding the UPS system in HD. It must be involved as it exists to degrade useless protein. However, the fact that it is unclear whether or not muHtt impairs the UPS means it is probably not as key to the pathogenesis of the disease as the mitochondria. More research is needed to determine the in vivo effects of muHtt on the UPS in HD neurons and if it is found to be comprehensively impaired, then models for cell death as a result of this should be considered. 5. Animal Models of Huntington’s DiseaseOne of the problems with understanding the pathogenic mechanisms of muHtt is the lack of a perfect animal model. Initial modelling efforts before the discovery of the IT15 gene used injections of glutamate receptor agonists to induce neurotoxicity and the subsequent loss of neurons in a manner similar to that seen in HD (McGeer & McGeer, 1976). While imperfect, these experiments showed potential pathways that were implicated in HD such as energetic defects associated with mitochondrial dysfunction. The major problem was the rapid onset of lesion formation, in contrast with the slow progression of HD. Clearly something different was needed. While the fact HD is caused by a defect in a single gene should suggest simplicity in generation of genetic models, the reality is much different. For a start, cloning the gene is difficult due to its size- plasmids can only contain inserted sequences of around 10-15kb (Htt is 170kb long), Taq polymerase used in cDNA generation has an error rate of around 1 per 1kb of DNA and finally there is no single muHtt protein due to the expanded nature of the polyQ region which varies dramatically between patients (Pouladi, Morton, & Hayden, 2013). Because of the varying nature of the gene, multiple approaches have been used in generating animal models (Fig. 7). These methods have resulted in the generation of C. elegans, D. melanogaster, rodent and other mammal models, all with their own strengths and weaknesses Both C. elegans and D. melanogaster are ultimately flawed because they do not display a Htt orthologue. However, expression of transgenic muHtt can be driven, leading to progressive degeneration and other similar symptoms to human HD, as well as a younger age of onset and with severity of symptoms proportional to the repeat length (Faber, Alter, Macdonald, & Hart, 1999)(Jackson, et al., 1998). Over 20 different rodent models are available, ranging from expression of exon 1 of human muHtt with 116 or 144 CAG repeats (R6/1 and R6/2 models respectively) to mice expressing endogenous mouse Htt under control of mouse promoter sequences. The latter are much more successful at expressing muHtt, with levels of expression equal to that of endogenous Htt in comparison to levels of 0.3-0.75x for R6/1 and R6/2 mice (Pouladi, Morton, & Hayden, 2013). For abnormally high levels of expression, different promoter sequences such as the cytomegalovirus (CMV) promoter can be used (Reddy, et al., 1999). Finally, large animal models are in development but show mixed success. Yang et al. (2008) transfected 130 Rhesus monkey oocytes with an 84 repeat length exon 1 of human Htt. Five pregnancies survived to term and expressed the desired gene, with four showing severe symptoms and one monkey being asymptomatic (Yang, et al., 2008). Unfortunately, the symptomatic monkeys all died with 6 months due to the extreme and rapid presentation of HD-like symptoms. This rapid onset was linked to possible overexpression of muHtt as in 3 of the 4 symptomatic monkeys it was found the gene had been inserted multiple times into the genome. While development of a non-human primate model would give the best animal model of HD, the rapid onset of symptoms and low yield of live births means continued research is needed. Attempts to generate sheep (Jacobsen, et al., 2010) and pig
models (Yang, et al., 2010) have encountered problems- the sheep model shows inclusion body pathology but a lack of an overt phenotype, whereas the pig model has a similar problem to the primate model in that few pregnancies were successful and animals died soon after birth. The reason for the massive variation in all types of animal models lies with the different methods for protein expression. Ideally, one would be able to state conclusively which method is the most effective but as some work exceptionally well in some animals and not at all in others, it appears that for the near future at least, researchers will have to pick a favoured model and stick to it in the development of treatments, hoping that results will translate to humans. 6. The Treatment of Huntington’s DiseaseThere is currently no cure for Huntington’s disease so current research looks into disease-modifying therapies. As it is clear muHtt is responsible for the pathogenesis of the disease, strategies include removal of toxic protein, prevention of aggregation, etc. Current clinical trials are also looking at the management of symptoms. The psychiatric symptoms can be relatively easily controlled with normal drugs in this field, whereas management of chorea, arguably the most severe of the symptoms, is still being researched. 6.1 The TRACK-HD Study and the Hurdles With Monitoring Disease ProgressionBefore the different treatment options are discussed, one crucial hurdle in conducting clinical trials is the lack of effective measurements to record progress or retardation of disease progression. Huntington’s progression is analysed by the Unified Huntington’s Disease Rating Scale (UHDRS), which assesses a number of specific markers (Huntington Study Group, 1996). The scale provides a number of assessments which the physician must rate from 0 to 4- as an example, arm rigidity, where 0 implies no rigidity and 4 implies severe. The overall rating of disease progression can then be provided as a total functional capacity score. The inherent problem with this is the reliance on a physician’s subjective opinion of symptoms. A recent, 24 month study called TRACK-HD aimed to rectify this by assessing a number of different biomarkers in a group of 349 people; 116 controls, 117 people carrying the muHtt gene but displaying no symptoms (premanifest HD) and 116 individuals showing early HD symptoms (Tabrizi, et al., 2012). By analysing biomarkers, physician bias can be eliminated and more accurate diagnoses and prognoses can be made. The TRACK-HD study found that was no abundance of markers to track disease progression but found that measuring striatal and total white-matter atrophy in the brain was useful. The lack of significant changes in cognitive, motor or oculomotor function recorded is probably due to the relatively short time period the study was carried out over- two years in a disease that takes decades to progress is not long. In addition, the most significant symptoms become apparent towards the end stage of the disease and no trial subjects were near this point; those with HD symptoms had only just begun to show them. Further biomarker studies are ongoing, with PREDICT-HD the next study likely to provide useful results (Biglan, et al., 2013). 6.2 Current Disease Management StrategiesAs chorea is the most debilitating symptoms sufferer’s tend to face, its management has been a key concern. The first drug to be approved for treatment of chorea by the FDA was tetrabenazine (TBZ), a catecholamine depleting agent. It promotes the early degradation of neurotransmitters such as dopamine, resulting in symptomatic relief from chorea (Chen, Ondo, Dashtipour, & Swope, 2012). Clinical trials have shown the drug to be useful in reducing symptoms. In a trial of 84 patients with HD showing chorea, 54 were given TBZ and 30 were given a placebo. It was found that the TBZ cohort had a statistically significant reduction (p<0.0001) in their UHDRS chorea score in comparison to those on the placebo (Huntington Study Group, 2006). However, the doses of TBZ administered to patients varied- doses were increased up to the maximum of 100mg a day until the desired outcome was noted, meaning the study is ineffective at providing a general dose for patients- dosing must instead be individually assessed. A further study investigated the withdrawal of TBZ over a 5 day period after administration, finding those who had complete withdrawal had a significant increase (p=0.0058) in their UHDRS chorea score of 5.3 units compared to those who stayed on the treatment or only had partial withdrawal (Frank, et al., 2008). While other antichoreic drugs such as amantadine, an antiparkinsonian, are being trialled after showing some positive effects (Lucetti, et al., 2002), generally the drugs prescribed aim to ‘reduce chorea to a level that is acceptable to the patient rather than to eliminate chorea completely’ (Frank, Treatment of Huntington's disease, 2014). Further, larger double blinded studies are needed to correctly analyse a drug’s efficacy. Treatment of psychiatric symptoms can be achieved through well-established drug therapies. Depression can be controlled using standard antidepressants such as fluoxetine (an SSRI) - this drug also has the added benefit of controlling irritability, another psychiatric symptoms displayed (Tyagi, Tyagi, Shekhar, Singh, & Kori, 2010). Other problems seen in HD include weight loss and dystonia, which can be managed by helpers ensuring patients are supported throughout their disease period. While recent trials have begun to show that some drugs may be useful in treating certain symptoms, there is a current lack of a unified treatment model. Instead, patients end up taking whichever drugs work best for them to control their individual symptoms. Because of this, the need to work on preventative therapies to slow progression of the disease and maybe even reverse its course is increasingly apparent. 6.3 Reduction in Pathogenic Protein Levels- Down regulation of the Mutant GeneAs it is clearly the mutant form of the Huntingtin protein that causes the damage to cells, if translation of mRNA into this protein can be lowered or even prevented this could potentially lead to a complete lack of symptoms. This has been the subject of multiple research papers. Blockade of muHtt fragment expression in transgenic mice between the age of 18 and 34 weeks revealed not only suppression of neurodegeneration but reversal of pathological features such as inclusion bodies (Yamamoto, Lucas, & Hen, 2000). This study used mice that had a promoter designed to specifically produce the muHtt fragment in the forebrain and could be switched off when necessary. While this shows that there may be a time period where changes in HD are reversible, the model does not translate well the human model of the disease- expression was induced by the researchers rather than being constant and the phenotypes progressed much more quickly.
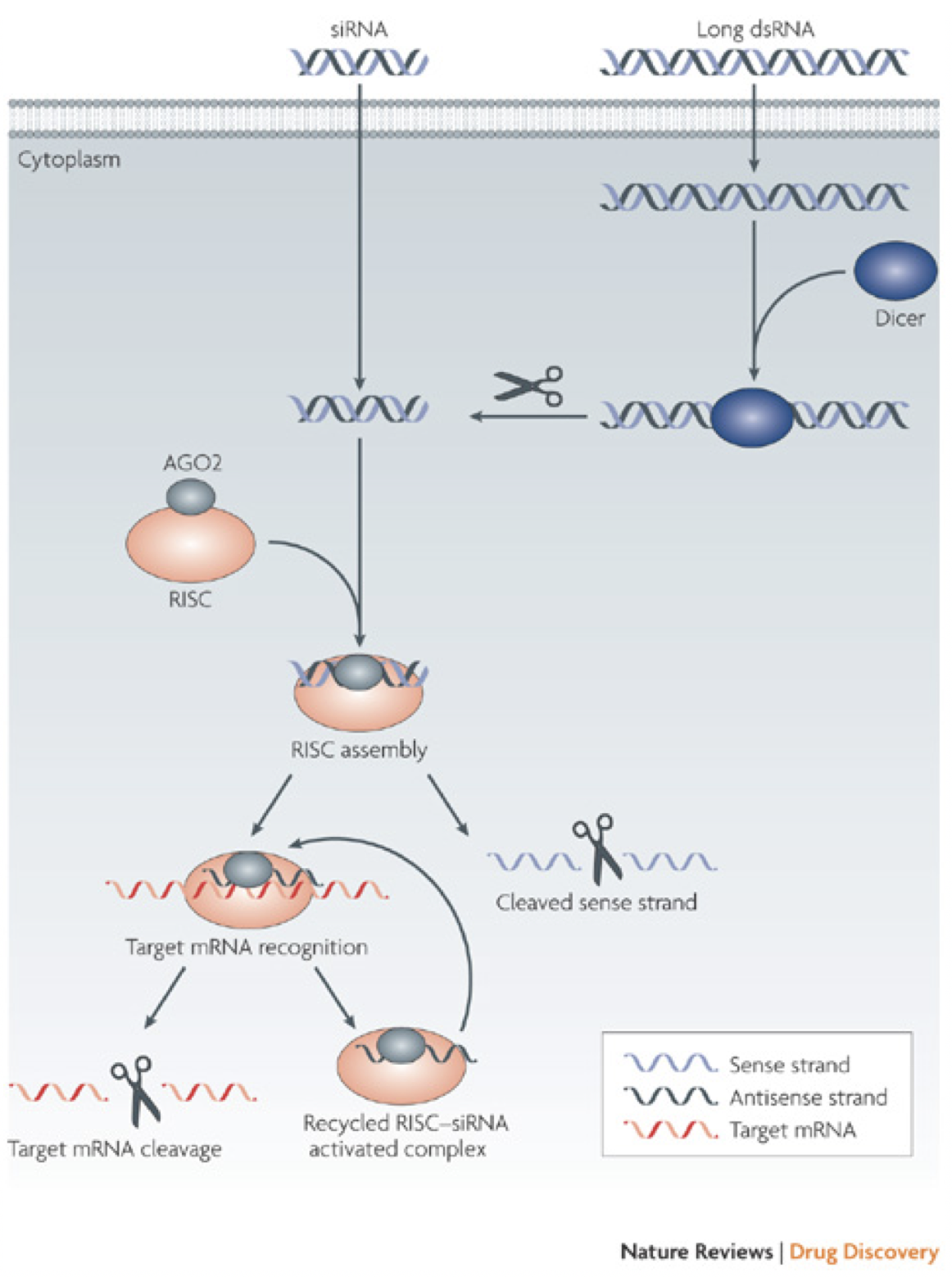
More recent research has looked into reducing muHtt expression in two ways. One of these is the use of RNA interference (RNAi), a process that uses small siRNA strands to bind to and inhibit translation or induce degradation of a complementary mRNA (Fig. 8). DiFiglia et al. (2007) introduced either a 100 CAG (mutant) or 18 CAG (wild type) repeat Htt cDNA strand into mouse brains using adeno-associated virus (AAV). Once HD-like symptoms were seen in the mutant but not wild type mice, either siRNA against Htt or control siRNA against luciferase was co-administered. The mice that received the siRNA-Htt showed significantly reduced mean inclusion body size in the striatum and significantly lowered aggregate numbers in the striatum. This change was associated with a reduction in phenotypic features of the disease (foot slip test and clasping test) (DiFiglia, et al., 2007). Once again however, there are flaws- the muHtt was effectively added to the mice rather than being endogenously expressed and the progression of symptoms was too fast to be related to human HD. Instead of using RNAi, antisense oligonucleotides could be used. Once again, these target the mRNA sequence of the Htt gene but differ from RNAi in that they have fewer off-target effects, reducing the impact of the treatment (Summerton, 2007). In vitro treatment of fibroblast cells derived from patients with either wtHtt or muHtt with oligonucleotides targeting the mutant sequence showed a reduction in Htt protein, an improvement in protection from neurotoxicity and no effect on other CAG repeat sequences around the body (e.g. TATA binding protein) (Hu, et al., 2009). Crucial criticisms of this study are the lack of an in vivo experiment to test relief of symptoms and the use of fibroblasts rather than neurons. Recent research used antisense oligonucleotides to induce exon skipping during mRNA maturation, after noting that exon 12 contained important cleavage sites for production of the toxic N-terminal fragment (Evers, et al., 2014). Replication of this procedure in a mouse model to see whether it ameliorates symptoms is clearly desirable in the future. One major problem with using RNAi or antisense oligonucleotides is the indiscriminate targeting of both muHtt and wtHtt, as reduction of wtHtt levels results in increased phenotype severity due to the loss of its protective, anti-apoptotic function (Zhang, et al., 2003). Targeting muHtt solely could be achieved through targeting SNP’s that are heterozygous to the mutant allele, rather than the expanded CAG repeat (Pfister & Zamore, 2009). 95% of HD chromosomes contain 22 specific SNPs that could be targeted by siRNA to prevent reduction of wtHtt levels (Warby, et al., 2009)(Lombardi, et al., 2009). This therapy could be useful to the majority of patients but some would lack the necessary SNPs for targeting. This could suggest a need to genotype every patient with HD and create personalised RNAi therapies. Reduction of levels of muHtt has an important role but further research is needed to create a treatment that is safe, specific and available for all patients at relatively low cost. Equally, the recent paper by Evers et al (2014) suggested the possibility that rather than completely removing the protein from cells, instead modification of the protein could be useful to prevent toxic fragment formation. 6.4 Aggregation Prevention- The Role of Molecular ChaperonesMolecular chaperones (heat shock proteins) are responsible for dealing with misfolded proteins within cells, by moving them to proteasomes for degradation. They also have roles in autophagy and apoptosis (Perrin, et al., 2007). It is likely that these roles are somehow impaired in the development of HD, as if the system was working correctly then muHtt would be dealt with without being able to form aggregates. This impairment is likely to be due to sequestration of the proteins into aggregate bodies before they can exert their effects. Attempts to rectify this process have used overexpression of the proteins to try to provide some restoration of function.
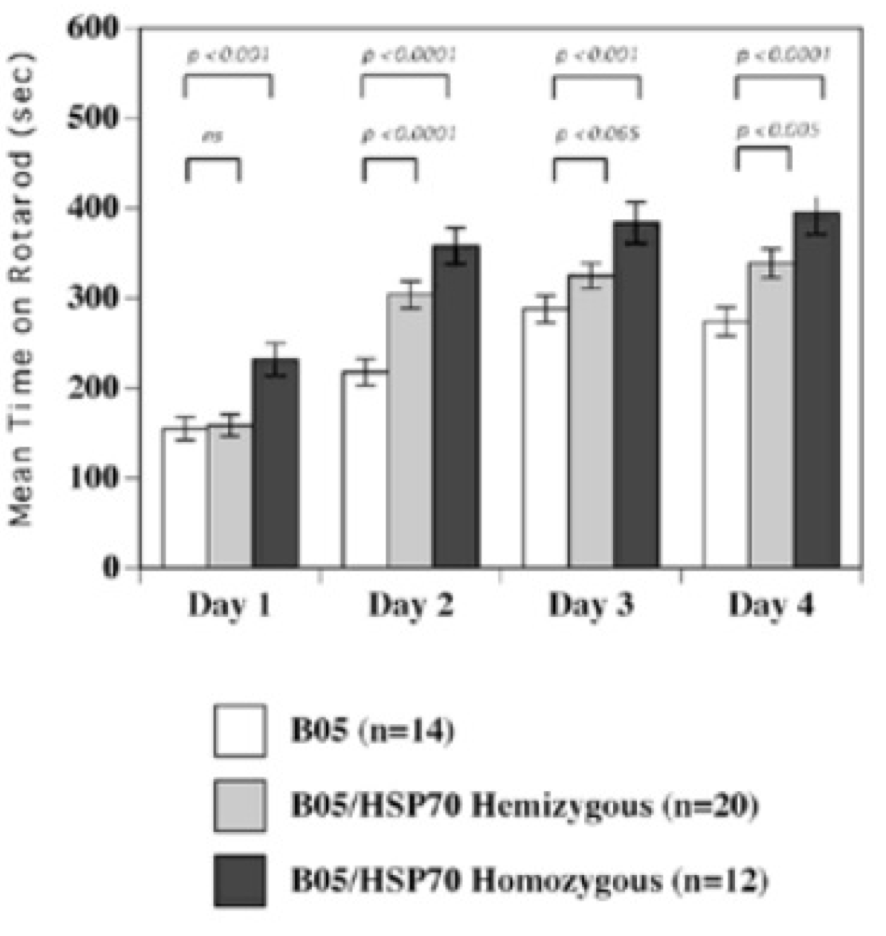
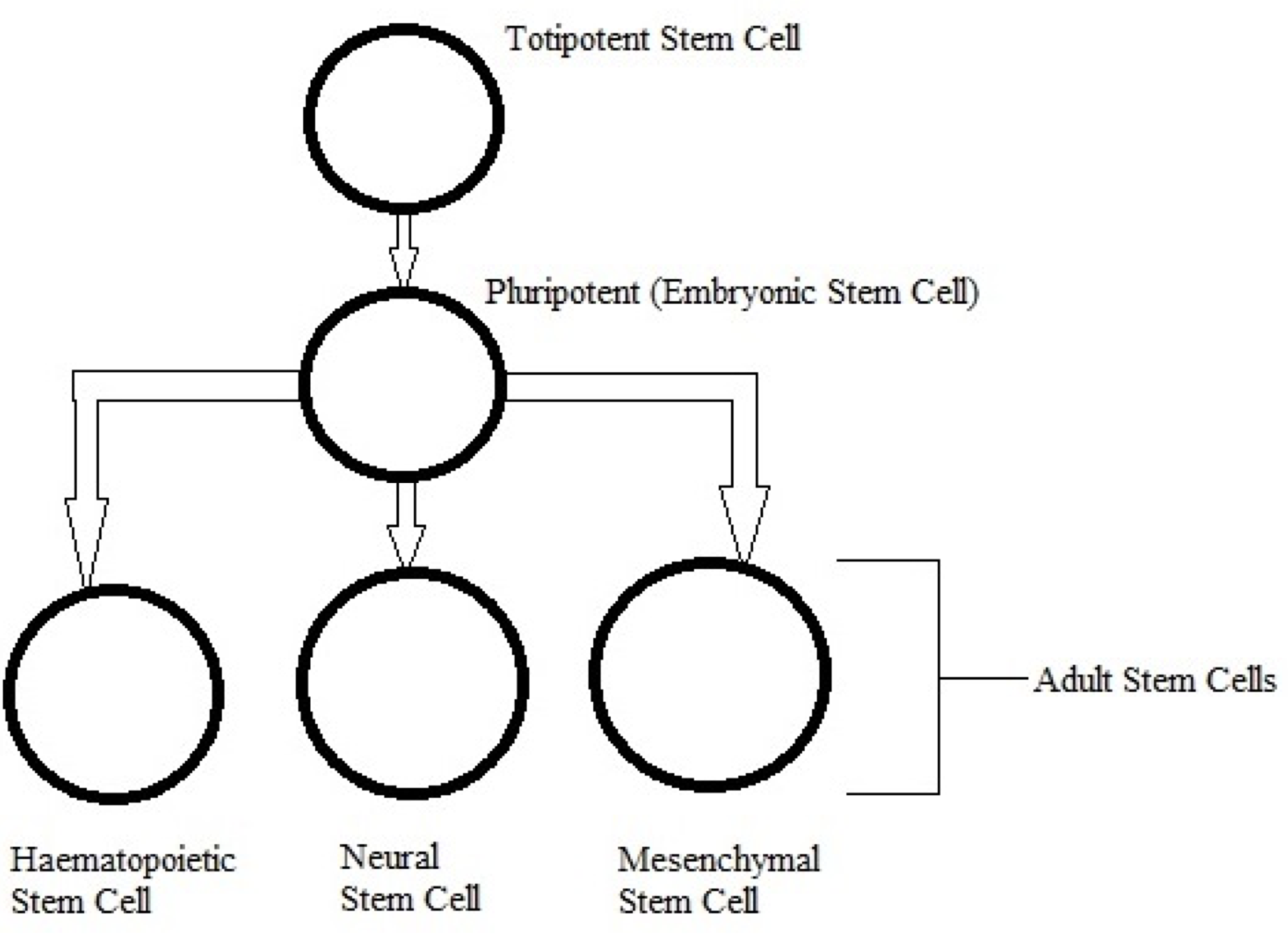
In mouse models, overexpression of chaperones can either have positive impacts (SCA1 mice, (Cummings, et al., 2001) (Fig. 9) or little to no effect (R6/2 mice, (Hansson, et al., 2003)). This indicates a need to find which chaperone is most useful for restoring function. The above studies used Hsp70 (70kDa), whereas usage of Hsp104 (104kDa) as a treatment in two different mice models of HD showed a reduction in aggregate formation and a reduction in muHtt toxicity (Perrin, et al., 2007)(Vacher, Garcia-Oroz, & Rubinsztein, 2005). However, one of the studies found no improvement in motor function. Until recently, it was unknown how the molecular mechanism involved in chaperone mediated treatment worked. Recently, it has been suggested that chaperones are involved in preventing aggregation of the N-terminal fragments rather than enhancing clearance of the full length mutant protein. If aggregates are formed by cross-linking the glutamines to other amino acids, this requires both a tissue transglutaminase and a source of amine groups (Violante, Luongo, Pepe, Annunziata, & Gentile, 2001). This source of amines can be from glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and it was shown that Hsp70 binds to both GAPDH and the polyQ region of toxic fragments, thereby preventing aggregation (Guzhova, et al., 2011). However, while this clearly suggests a role for chaperones in aggregation prevention, this is not necessarily a good thing as it is unknown whether aggregates and the subsequent inclusion bodies are a protective function of the cell against HD, or a pathogenic mechanism that contributes to cell death. The fact that overexpression of different chaperones in different mouse models results in either positive or negative outcomes further adds to this confusion- by preventing formation of aggregates, this would result in raised levels of toxic soluble fragments. Chaperones could, however, be used in conjunction with another therapy; for example, chaperones-induced raised levels of soluble fragments could then be targeted by a novel drug. There is potential for future research but whether or not chaperones are useful in the prevention remains unclear. 6.5 Replacement of Lost Neurons- Stem Cell TherapyArguably the most promising future therapies for many diseases including HD is the use of stem cells to regenerate tissue. Stem cells are undifferentiated cells which have the ability, when exposed to various conditions and growth factors, to differentiate into specialised cells. If stem cells could be introduced to the brains of HD patients and forced to replace lost neurons, this could reduce the phenotype and possibly prevent the disease. Stem cells are divided into two categories (Fig. 10); adult stem cells, which have a limited differentiating ability and embryonic stem cells (eSC) which have the ability to commit to multiple lineages. Neural stem cells (NSCs) would seem to be the most obvious initial choice for a HD therapy. Initial research looked at using injections of NSCs into rat models of HD that used the early model mechanism of injecting quinolinic acid to induce lesion formation (Ryu, et al., 2004). They found that mice given an injection of NSCs prior to induction of lesion formation had reduced amounts of striatal damage and improved motor function. However, injection of NSCs after lesion formation had no restorative effect. This suggests that stem cells can be protective but not curative and ultimately the research was flawed as the rat model used has rapid, acute lesion formation in comparison to the slow progression of human HD.
Usage of NSCs in a transgenic rat model of HD with a 51 CAG repeat Htt gene under rat promoter control has been studied. 12 month old rats with HD symptoms were given transplantations of NSCs and for the first 4 weeks following this, motor symptoms were reduced (Rossignol, et al., 2014). However, 6 weeks after transplant the rats began to show motor defects similar to the controls, suggesting that long-term therapy with NSCs may not be possible. However, this same study looked at the usage of mesenchymal stem cells (MSCs) for transplant instead of NSCs, finding that MSC transplants provided a longer term protective effect. This agrees with findings from lesion inducing rat models (Rossignol, et al., 2011). The reason for the short term effect is probably due to an inflammatory reaction and immune response to the transplant site. Other non-neural stem cells have been tested with positive results. Adipose-derived stem cells (ASCs, a form of MSC) have been shown to have neuroprotective effects in both quinolinic acid models and R6/2 models, along with improved motor function and a reduction in aggregate levels (Lee, et al., 2009)(Im, et al., 2013). Im et al. attributed this to restoration of PGC1α function, a protein involved in mitochondrial function and down regulated in HD as it is under the CREB control, which is sequestered by muHtt (McGill & Beal, 2006). Non-neural stem cells could also be contributing to reduced pathogenesis due to their secretion of various growth factors, including BDNF, which has been shown to stimulate new neuron growth and promote healing of damaged nerves(Lopatina, et al., 2011) (Wilkins, et al., 2009). There is huge potential in the use of stem cells in HD but this seems to be solely preventative with no curative effects- while some stem cells do induce the growth of new neural tissue, this tissue will still produce muHtt and resultantly will still be eventually destroyed. The short life span of stem cell grafts would mean sufferers needing regular transplants. As HD takes so long to progress in most cases, the ideal scenario would be a combination treatment, using stem cells to regenerate tissue along with RNAi to knock down muHtt and hopefully extend the cell lifespan, to the point where symptoms take so long to appear that the patient can live a normal life. While using neural or mesenchymal stem cells for treatment has been tested, there has been no research into using either embryonic stem cells or induced pluripotent stem cells. Instead, current research is looking at using these types of cells to generate an in vitro human model of HD obviating the need to use actual patients. Using HD embryos, researchers managed to produce to embryonic stem cell lineages expressing a 37 CAG repeat and a 51 CAG repeat, which went on to express mRNA when cultured (Niclis, et al., 2009). Using the signalling protein Noggin, the stem cells formed neurospheres; cell cultures of free neural stem cells. Unfortunately, the cells were not found to contain inclusion bodies, suggesting the model needs further refinement. iPSCs have also been used to generate cell models of HD. Unlike eSCs, iPSCs are generated from adult cells, meaning there is no need to worry about the ethical problems of using embryos. By adding defined transcription factors (Yu, et al., 2007) to human fibroblasts, they produced iPSCs which were then differentiated into NSCs (HD iPSC Consortium, 2012). These NSCs displayed expanded CAG repeats, produced muHtt mRNA and were more susceptible to HD related cell death. Both eSCs and iPSCs are showing promise for a stable, human model of HD, overcoming the problems with different animal models previously discussed. 6.6 Other Potential TherapiesAs mitochondrial dysfunction and subsequent susceptibility to excitotoxicity are well characterised in HD, prevention of this may rescue cells from death. NMDA receptors, which bind glutamate and open to allow calcium influx, can be antagonised by drugs such as amantadine (Kornhuber, Weller, Schoppmeyer, & Riederer, 1994). Unfortunately, amantadine has been trialled in humans with little success- patients reported they felt better, but showed no symptomatic improvements (O'Suilleabhain & Dewey, 2003). Other antagonists such as memantine may slow progression of HD (Beister, et al., 2004), but one of the problems with using this drug class is multiple effects around the body. Finally, caspase inhibition to prevent cleavage of muHtt and the subsequent build-up of toxic fragments could be useful, having been shown to reduce neuron loss in mice (Ona, et al., 1999). This treatment must be studied in the long term inhibition of caspases can lead to tumour growth and subsequent resistance to chemotherapy (Fischer, Janssen, & Schulze-Osthoff, 2007).Continued on Next Page » 
From the Inquiries Journal Blog")   Related ReadingMonthly Newsletter SignupThe newsletter highlights recent selections from the journal and useful tips from our blog. Suggested Reading from Inquiries Journal
Inquiries Journal provides undergraduate and graduate students around the world a platform for the wide dissemination of academic work over a range of core disciplines. Representing the work of students from hundreds of institutions around the globe, Inquiries Journal's large database of academic articles is completely free. Learn more | Blog | Submit Follow IJ
Latest in Neuroscience |