Are E-Cigarettes a Safe Alternative to Smoking? Using Airway Cell Cultures to Assess Early Indicators of COPD Progression
2015, Vol. 7 No. 05 | pg. 1/2 | »
Abstract
There is limited data available on the safety of electronic (E)-cigarettes, despite their commercial availability and widespread use. E-cigarettes are electronic devices that deliver nicotine to the lungs but limited data is available about their impact on lung health. This study attempts to address the potential health effects of E-cigarettes utilizing airway cell culture techniques to assess cellular toxicity and effects on protein activation and the induction of responses associated with chronic obstructive pulmonary disease (COPD). Currently, COPD is the third highest cause of death in the USA and tobacco smoke use is the primary cause of disease formation. The liquid found in E-cigarettes (E-liquid) was found to be toxic to lung cells even when diluted to very low levels and activated similar proinflammatory kinases as tobacco-generated cigarette smoke. These activated kinases could potentially lead to lung inflammation and increased protease activity, which are both associated with COPD progression. Additionally, E-liquid appeared to increase the activation of NF-kappaB, a specific target of kinases. Based on these findings, this study concludes that E-cigarette usage has the potential to induce inflammation in airway cells and lead to harmful effects similar to those observed in COPD.
In an effort to find a safer replacement for smoking, many people have turned to E-cigarettes, which are cylindrical devices that have a strikingly similar appearance to conventional cigarettes. E-cigarettes are comprised of a battery and a replaceable cartridge with a propylene glycol or glycerin liquid solution containing differing quantities of nicotine. The liquid solution vaporizes with the heat of the battery, and enters the lung upon inhalation. In contrast to traditional cigarettes, this apparatus in theory releases far fewer toxicants by eliminating the smoke component of cigarettes [8, 9]. However despite their widespread use, the health effects of E-cigarettes and their exposure products have yet to be fully investigated, especially with regard to chronic obstructive pulmonary disease (COPD) progression.
{kind=link}
Advertisement
COPD is a progressive disease that induces airflow obstruction thereby making it increasingly difficult to breathe [3]. Over time, there is a loss of elasticity in the lung due to oxidative stress, apoptosis and protease expression [4]. COPD is currently the third leading cause of death in the United States [3] and is estimated to develop into the third leading cause of death globally within the next twenty years [5].
Over the past thirty years, the age-adjusted mortality rate correlated with COPD has grown at an alarming rate of 71% despite readily available pharmacological treatments [3]. Cigarette smoke is a primary inducer of COPD, as most people afflicted are either current or former smokers [3]. There are projected to be more than a billion tobacco users on a global scale [7]. Accordingly, more effective treatment strategies are needed to directly address the fundamental disease pathogenesis and the issue of smoking.
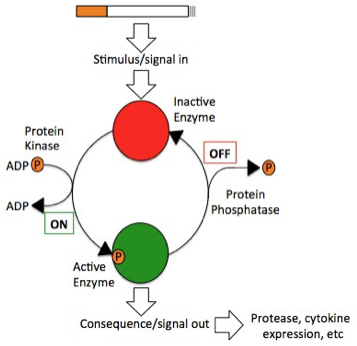
This study attempted to test the toxicity of E- cigarettes using epithelial A549 lung cells, a residing lung cell type that would be exposed to both cigarette smoke and E-cigarette vapor in-vitro. Additionally, this project attempted to determine whether the nicotine was the primary factor in E-cigarettes that could activate specific targets and transcription factors associated with inflammatory signaling responses within A549 (lung epithelial) cells and COPD. Cigarette smoke induces phosphorylation, and, thus, activation, of key proteins associated with inflammation (Figure 1).
Methodology
Cell Culture
Human A549 cells (ATCC® CCL-185™, ATCC, Manassas, VA) were grown to 70% confluency on Dulbecco’s modified Eagle Medium (gibco® DMEM(1X) + GlutaMAXTM-1) treated with Fetal Bovine Serum and Penicillin/Strep. Media was replaced every 2 days during the growth period. After 70% confluence was achieved media was drained, cells were rinsed with PBS, and trypsin (enzyme) was added. The resulting solution of cells and trypsin was transferred to a test tube and centrifuged for 5 minutes at 1100 rpm at room temperature. Cells were then re-suspended into fresh media and 2 ml of the resulting solution was pipetted into each well of a 96-well plate.
Cells were allowed to adhere to the flask overnight. Once cells had reached 60-80% confluence, they were used for experiments with E-liquid. We exposed A549 epithelial lung cells to various concentrations of nicotine alone (0.01 mM) or E-liquid (American eLiquidTM; 50:50 v/v propylene glycol to vegetable glycerin) with or without nicotine (100% dose has 36 mg/ml (or 222 mM) of nicotine). E-liquid was diluted in culture media to the concentrations outlined in the figures.
Figure 2. Design for standard 96-well plate used in the Lactic Dehydrogenase Assay.
{kind=link}
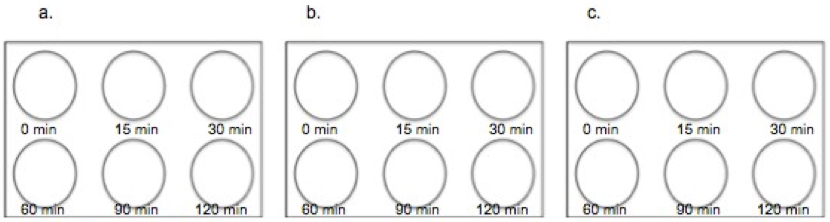
Figure 3. An example of the experimental plate and concentrations of E-liquid used in the subtoxic assays. A, B, and C represent standard, 6-well plates containing A549 cells. Each plate was treated once with 1% (2.2 mM) E-liquid and samples were collected at the following time points: 0, 15, 30, 60, 90, and 120 minutes.
{kind=link}
LDH Assay
To determine toxic doses of E-liquid, A549 cells were treated with 2 mL of the following concentrations of E-liquid minus nicotine: 0, 0.22, 2.2, 11, 22 & 44 mM (0, 0.1, 1, 5, 10 & 20%, respectively). PBS was used as a negative control. Cell death was colormetrically assessed by quantifying Lactic Dehydrogenase (LDH) release via the LDH Assay. Media was collected from cells 24 hours after performing the exposures listed above and LDH release was determined in this media.
Advertisement
Using a 96-well plate, 50 µL of media from each condition was examined in triplicate (Figure 2). A 5 ml mixture containing 1666.7ul of each LDH assay substrate solution, LDH assay cofactor, and LDH assay dye solution was prepared, of which 50 µL (standard kit volume) was pipetted into each well containing samples (Figure 2).
The plate was then incubated in the dark for 30 minutes, and substrate absorbance was measured at 410nm using a Tecan Genios spectrophotometer. The rate of substrate turnover is measured by change in absorbance and is proportional to LDH release and is corresponds to cellular stress due to toxicity.
E-Liquid Time Study
After performing the LDH Assay, it was determined that 2.2 mM E-liquid was an appropriate sub-lethal dose for assessment of kinase activation. The effects of 1% E-cigarette liquid on MAP kinase signaling were examined. A549 cells were treated once with 1% E-liquid and samples were collected at the following time points: 0, 15, 30, 60, 90, and 120 minutes (Figure 3). The E-liquids (with or without 2.2 mM nicotine) (50%PG/50%VG) solutions were diluted to a final concentration of 1%, which corresponded to 2,2mM nicotine. Cells were treated as outlined in Figure 3.
After 120 minutes, wells were drained of media and washed with PBS. 150 µL of broad range protease inhibitor solution (Fisher Scientific) (27 µl of protease inhibitors, 27 µL of EDTA, and 2700 µL of lysis buffer RIPA) was pipetted into each well. The RIPA buffer caused the cells to burst so that the protein could be readily extracted. These protein samples were stored on ice throughout the extraction process.
Protein Expression
Western blots. The effects of E-liquid on MAP kinase signaling were examined by performing Western blots for the following targets: p-Erk, p p-38, p-JNK, p38, JNK, ERK and Actin. 10 µL of the extracted protein samples were mixed with 3 µL of a 5X sample buffer in microcentrifugation tubes, heated to 90oC for 5 minutes, centrifuged (2,000 x g for 1 minute) and gel electrophoresis took place at 100 Volts for 60 minutes at room temperature. The proteins were extracted and transferred to a nitrocellulose membrane through via three different methods: Semi- dry transfer machine (Trans-Blot® SD Semi-Dry Transfer Cell, Bio-Rad), Wet transfer machine (Wet/Tank Blotting Systems, Bio-Rad), and Turbo-transfer machine (Trans-Blot® Turbo™ Transfer Starter System, Bio-Rad).
The nitrocellulose membrane was rinsed in distilled water, Sigma® Ponceau S distilled water again, and then placed in a 5% milk solution containing 2.5 µl of lactose powder (Fisher Scientific) and 47.5 µl of PBS solution for one hour. The primary antibody was prepared using 4 µl of the given antibody solution (p-Erk, p p-38, p-JNK, p38, JNK, ERK and Actin) and 4 ml of a 2% BSA (Bovine Serum Albumin) Solution. The nitrocellulose membrane was placed in the primary antibody solution and left to incubate overnight with mild agitation (gentle rocking). The next morning, the nitrocellulose membrane was removed from the primary antibody solution and rinsed in a wash buffer solution three times for 10 minutes each. The nitrocellulose membrane was placed in the prepared second antibody solution containing 4 µL of Anti-Rabbit HRP solution and 4 ml of a 5% milk solution for two hours. For the phosphorylated proteins, 1,000 µl each of SuperSignal® West Femto Luminol/Enhancer Solution (Thermo Scientific) and SuperSignal® West Femto Stable Peroxide Buffer (Thermo Scientific) were mixed and pipetted onto the nitrocellulose membrane.
For the total proteins, 500 µl each of SuperSignal® West Pico Luminol Enhancer Solution (Thermo Scientific), SuperSignal® West Pico Stable Peroxide Solution (Thermo Scientific, SuperSignal® West Femto Luminol/Enhancer Solution (Thermo Scientific) and SuperSignal® West Femto Stable Peroxide Buffer (Thermo Scientific) were mixed and pipetted onto the nitrocellulose membrane. After five minutes, the nitrocellulose membrane was placed in the Bio Rad Gel Dock Station (ChemiDoc™ XRS+ System, Bio-Rad). A chemi-luminescence camera was used to detect the concentration of Anti-Rabbit Horseradish Peroxidase HRP (secondary antibody) proportional to the amount of protein present.
Densitometry Analysis of Proteins. The proteins on the nitrocellulose membrane were analyzed through densitometry analysis (Figure 4). Densitometry was performed on each target and represented as a ratio of pixel intensity compared to actin, using Bio-Rad Laboratories Image Lab software (version 4.0, build 16).
This technique was used to quantify the level of activation of proteins by calculating a ratio of phosphorylated protein to total protein. The ratio was proportional to the level of activation of the protein. Graph Pad Prism was used to create a densitometry graph. Results were analyzed using Graph Pad Prism and a T-test, and the statistically significant difference was reported to be any value less than 0.05.
Figure 4. An example schematic diagram of the densitometry analysis
Detecting Transcription Factor Activation
Nuclear and cytoplasmic protein fractionation. Nuclear and cytoplasmic protein fractionation was performed to extract nuclear protein to detect the phosphorylation of NF-kappaB following E-liquid treatment. The treated cells from the 96-well plates were washed in ice-cold PBS. Cells were removed and lysed by scraping in 150 µl of cytoplasmic fraction buffer. Cytoplasmic fraction was then incubated at 4 °C for 30 minutes (rotating). Cytoplasmic fraction was centrifuged at 10,000 rpm for 10 minutes at 4 °C and supernatant liquid (cytoplasmic extract) was removed.
The pellet remaining at the bottom of the tube was washed with 1 ml PBS to remove any remaining cytoplasmic contents. Cell fraction was centrifuged again at 10,000 rpm for 10 minutes at 4 °C, and PBS was removed. The remaining nuclear pellet was lysed with 70 µl of nuclear buffer and incubated at 4 °C for 30 minutes (rotating). The nuclear solution was centrifuged at 14,000 rpm for 10 minutes at 4 °C. The supernatant (nuclear extract) was then removed and stored at -80 °C until required for use. The nuclear pellet was discarded.
Transcription factor assay to detect activation of NF-kappaB and AP-1. TransAMTM NF-kappaB p65 (Active Motif) Assay Kit was used to detect and quantify transcription factor (NF-kappaB and AP-1) activation. Simply, cell extract containing activated transcription factor was added to the oligonucleotide-coated plate. Using antibodies directed against the NF-kappaB/AP-1 p65 or c-jun AP-1 subunits and horseradish peroxidase (HRP), a secondary antibody, activated NF-kappa B or AP-1 was detected colorimetrically using a spectrophotometer. The amount of DNA binding was considered proportional to the amount of NF-kappaB activity. We did this by first adding 30µl Complete Binding Buffer and 20uL nuclear extract sample diluted in 1 mL of lysis buffer to each well. 1 µL Jurkat Nuclear extract (positive control of nuclear extract with active Nf-kappaB supplied with this kit) in 19 µL of Complete Lysis Buffer was added as the positive control. 15 µL Lysis buffer was added as the negative control.
The plate was then incubated for one hour at room temperature with mild agitation (100 rpm on a rocking platform). Each well was washed three times with 200 µL 1X Wash Buffer. 100 µL of diluted NF-kappaB antibody (1:1,000 dilution in 1X Antibody Binding Buffer) was added to each of the 32 wells. The plate was covered and incubated for one hour at room temperature without agitation. The wells were washed three times with 200 µL 1X Wash Buffer, as described previously. 100 µL of diluted HRP-conjugated antibody (1:1,000 dilution in 1X Antibody Binding Buffer) was added to all 32 wells. The plate was covered and incubated for one hour at room temperature without agitation.
The wells were washed four times with 200 µL 1X Wash Buffer. 100 µL Developing Solution was added to all 32 wells. The plate was incubated for five minutes at room temperature protected from direct light until the sample wells turned a medium to dark blue. 100 µL Stop Solution was added. In the presence of the acid, the blue color turned yellow. Absorbance was read on a Tecan Genios spectrophotometer within five minutes at 450 nm.Continued on Next Page »